U.S. Department of Health and Human Services
Food and Drug Administration
Center for Drug Evaluation and Research (CDER)
Center for Biologics Evaluation and Research (CBER)
June 2006
ICH
TABLE OF CONTENTS
I. INTRODUCTION (1)
II. SCOPE (2)
III. PRINCIPLES OF QUALITY RISK MANAGEMENT (3)
IV. GENERAL QUALITY RISK MANAGEMENT PROCESS (4)
A. Responsibilities (4.1)
B. Initiating a Quality Risk Management Process (4.2)
C. Risk Assessment (4.3)
D. Risk Control (4.4)
E. Risk Communication (4.5)
F. Risk Review (4.6)
V. RISK MANAGEMENT METHODOLOGY (5)
VI. INTEGRATION OF QUALITY RISK MANAGEMENT INTO INDUSTRY AND REGULATORY OPERATIONS
VII. DEFINITIONS (7)
VIII. REFERENCES (8)
ANNEX I: RISK MANAGEMENT METHODS AND TOOLS
I.1 Basic Risk Management Facilitation Methods
I.2 Failure Mode Effects Analysis (FMEA)
I.3 Failure Mode, Effects and Criticality Analysis (FMECA)
I.5 Hazard Analysis and Critical Control Points (HACCP)
I.6 Hazard Operability Analysis (HAZOP)
I.7 Preliminary Hazard Analysis (PHA)
ANNEX II: POTENTIAL APPLICATIONS FOR QUALITY RISK MANAGEMENT
II.1 Quality Risk Management as Part of Integrated Quality Management
II.2 Quality Risk Management as Part of Regulatory Operations
II.3 Quality Risk Management as Part of Development
II.4 Quality Risk Management for Facilities, Equipment and Utilities
II.5 Quality Risk Management as Part of Materials Management
II.6 Quality Risk Management as Part of Production
II.7 Quality Risk Management as Part of Laboratory Control and Stability Studies
II.8 Quality Risk Management as Part of Packaging and Labelling
Guidance for Industry[1]
Q9 Quality Risk Management
This guidance represents the Food and Drug Administration's (FDA's) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance.
I. INTRODUCTION (1)[2]
Risk management principles are effectively utilized in many areas of business and government including finance, insurance, occupational safety, public health, pharmacovigilance, and by agencies regulating these industries. Although there are some examples of the use of quality risk management in the pharmaceutical industry today, they are limited and do not represent the full contributions that risk management has to offer. In addition, the importance of quality systemshas been recognized in the pharmaceutical industry, and it is becoming evident that quality risk management is a valuable component of an effective quality system.
It is commonly understood that risk is defined as the combination of the probability of occurrence of harm and theseverity of that harm. However, achieving a shared understanding of the application of risk management among diversestakeholders is difficult because each stakeholder might perceive different potential harms, place a different probability on each harm occurring and attribute different severities to each harm. In relation to pharmaceuticals, although there are a variety of stakeholders, including patients and medical practitioners as well as government and industry, the protection of the patient by managing the risk to quality should be considered of prime importance.
The manufacturing and use of a drug product, including its components, necessarily entail some degree of risk. The risk to its quality is just one component of the overall risk. It is important to understand that product quality should be maintained throughout the product lifecycle such that the attributes that are important to the quality of the drug product remain consistent with those used in the clinical studies. An effective quality risk management approach can further ensure the high quality of the drug product to the patient by providing a proactive means to identify and control potential quality issues during development and manufacturing. In addition, use of quality risk management can improve the decision making if a quality problem arises. Effective quality risk management can facilitate better and more informed decisions, can provide regulators with greater assurance of a company’s ability to deal with potential risks, and can beneficially affect the extent and level of direct regulatory oversight.
The purpose of this document is to offer a systematic approach to quality risk management. It serves as a foundation or resource document that is independent of, yet supports, other ICH Quality documents and complements existing quality practices, requirements, standards, and guidelines within the pharmaceutical industry and regulatory environment. It specifically provides guidance on the principles and some of the tools of quality risk management that can enable more effective and consistent risk-based decisions, by both regulators and industry, regarding the quality of drug substances and drug products across the product lifecycle. It is not intended to create any new expectations beyond the current regulatory requirements.
It is neither always appropriate nor always necessary to use a formal risk management process (using recognized tools and/or internal procedures, e.g., standard operating procedures). The use of informal risk management processes (using empirical tools and/or internal procedures) can also be considered acceptable. Appropriate use of quality risk management can facilitate but does not obviate industry’s obligation to comply with regulatory requirements and does not replace appropriate communications between industry and regulators.
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
II. SCOPE (2)
This guidance provides principles and examples of tools for quality risk management that can be applied to different aspects of pharmaceutical quality. These aspects include development, manufacturing, distribution, inspection, and submission/review processes throughout the lifecycle of drug substances, drug products, biological and biotechnological products (including the use of raw materials, solvents, excipients, packaging and labeling materials in drug products, biological and biotechnological products).
III. PRINCIPLES OF QUALITY RISK MANAGEMENT (3)
Two primary principles of quality risk management are:
· The evaluation of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient; and
· The level of effort, formality, and documentation of the quality risk management process should be commensurate with the level of risk.
IV. GENERAL QUALITY RISK MANAGEMENT PROCESS (4)
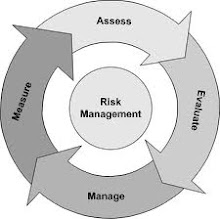
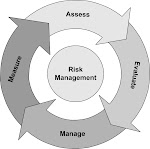
Quality risk management is a systematic process for the assessment, control, communication and review of risks to the quality of the drug product across the product lifecycle. A model for quality risk management is outlined in the diagram (Figure 1). Other models could be used. The emphasis on each component of the framework might differ from case to case but a robust process will incorporate consideration of all the elements at a level of detail that is commensurate with the specific risk.
Figure 1: Overview of a typical quality risk management process

Decision nodes are not shown in the diagram above because decisions can occur at any point in the process. These decisions might be to return to the previous step and seek further information, to adjust the risk models or even to terminate the risk management process based upon information that supports such a decision. Note: “unacceptable” in the flowchart does not only refer to statutory, legislative, or regulatory requirements, but also to indicate that the risk assessment process should be revisited.
A. Responsibilities (4.1)
Quality risk management activities are usually, but not always, undertaken by interdisciplinary teams. When teams are formed, they should include experts from the appropriate areas (e.g., quality unit, business development, engineering, regulatory affairs, production operations, sales and marketing, legal, statistics, and clinical) in addition to individuals who are knowledgeable about the quality risk management process.
Decision makers should
· take responsibility for coordinating quality risk management across various functions and departments of their organization and
· ensure that a quality risk management process is defined, deployed, and reviewed and that adequate resources are available.
B. Initiating a Quality Risk Management Process (4.2)
Quality risk management should include systematic processes designed to coordinate, facilitate and improve science-based decision making with respect to risk. Possible steps used to initiate and plan a quality risk management process might include the following:
· Define the problem and/or risk question, including pertinent assumptions identifying the potential for risk
· Assemble background information and/or data on the potential hazard, harm or human health impact relevant to the risk assessment
· Identify a leader and critical resources
· Specify a timeline, deliverables, and appropriate level of decision making for the risk management process
C. Risk Assessment (4.3)
Risk assessment consists of the identification of hazards and the analysis and evaluation of risks associated with exposure to those hazards (as defined below). Quality risk assessments begin with a well-defined problem description or risk question. When the risk in question is well defined, an appropriate risk management tool (see examples in section 5) and the types of information that will address the risk question will be more readily identifiable. As an aid to clearly defining the risk(s) for risk assessment purposes, three fundamental questions are often helpful:
1. What might go wrong?
2. What is the likelihood (probability) it will go wrong?
3. What are the consequences (severity)?
Risk identification is a systematic use of information to identify hazards referring to the risk question or problem description. Information can include historical data, theoretical analysis, informed opinions, and the concerns of stakeholders. Risk identification addresses the “What might go wrong?” question, including identifying the possible consequences. This provides the basis for further steps in the quality risk management process.
Risk analysis is the estimation of the risk associated with the identified hazards. It is the qualitative or quantitative process of linking the likelihood of occurrence and severity of harms. In some risk management tools, the ability to detect the harm (detectability) also factors in the estimation of risk.
Risk evaluation compares the identified and analyzed risk against given risk criteria. Risk evaluations consider the strength of evidence for all three of the fundamental questions.
In doing an effective risk assessment, the robustness of the data set is important because it determines the quality of the output. Revealing assumptions and reasonable sources of uncertainty will enhance confidence in this output and/or help identify its limitations. Uncertainty is due to combination of incomplete knowledge about a process and its expected or unexpected variability. Typical sources of uncertainty include gaps in knowledge, gaps in pharmaceutical science and process understanding, sources of harm (e.g., failure modes of a process, sources of variability), and probability of detection of problems.
The output of a risk assessment is either a quantitative estimate of risk or a qualitative description of a range of risk. When risk is expressed quantitatively, a numerical probability is used. Alternatively, risk can be expressed using qualitative descriptors, such as “high,” “medium,” or “low,” which should be defined in as much detail as possible. Sometimes a risk score is used to further define descriptors in risk ranking. In quantitative risk assessments, a risk estimate provides the likelihood of a specific consequence, given a set of risk-generating circumstances. Thus, quantitative risk estimation is useful for one particular consequence at a time. Alternatively, some risk management tools use a relative risk measure to combine multiple levels of severity and probability into an overall estimate of relative risk. The intermediate steps within a scoring process can sometimes employ quantitative risk estimation.
D. Risk Control (4.4)
Risk control includes decision making to reduce and/or accept risks. The purpose of risk control is to reduce the risk to an acceptable level. The amount of effort used for risk control should be proportional to the significance of the risk. Decision makers might use different processes, including benefit-cost analysis, for understanding the optimal level of risk control.
Risk control might focus on the following questions:
· Is the risk above an acceptable level?
· What can be done to reduce or eliminate risks?
· What is the appropriate balance among benefits, risks and resources?
· Are new risks introduced as a result of the identified risks being controlled?
Risk reduction focuses on processes for mitigation or avoidance of quality risk when it exceeds a specified (acceptable) level (see Fig. 1). Risk reduction might include actions taken to mitigate the severity and probability of harm. Processes that improve the detectability of hazards and quality risks might also be used as part of a risk control strategy. The implementation of risk reduction measures can introduce new risks into the system or increase the significance of other existing risks. Hence, it might be appropriate to revisit the risk assessment to identify and evaluate any possible change in risk after implementing a risk reduction process.
Risk acceptance is a decision to accept risk. Risk acceptance can be a formal decision to accept the residual risk or it can be a passive decision in which residual risks are not specified. For some types of harms, even the best quality risk management practices might not entirely eliminate risk. In these circumstances, it might be agreed that an appropriate quality risk management strategy has been applied and that quality risk is reduced to a specified (acceptable) level. This (specified) acceptable level will depend on many parameters and should be decided on a case-by-case basis.
E. Risk Communication (4.5)
Risk communication is the sharing of information about risk and risk management between the decision makers and others. Parties can communicate at any stage of the risk management process (see Fig. 1: dashed arrows). The output/result of the quality risk management process should be appropriately communicated and documented (see Fig. 1: solid arrows). Communications might include those among interested parties (e.g., regulators and industry; industry and the patient; within a company, industry, or regulatory authority). The included information might relate to the existence, nature, form, probability, severity, acceptability, control, treatment, detectability, or other aspects of risks to quality. Communication need not be carried out for each and every risk acceptance. Between the industry and regulatory authorities, communication concerning quality risk management decisions might be effected through existing channels as specified in regulations and guidances.
F. Risk Review (4.6)
Risk management should be an ongoing part of the quality management process. A mechanism to review or monitor events should be implemented.
The output/results of the risk management process should be reviewed to take into account new knowledge and experience. Once a quality risk management process has been initiated, that process should continue to be utilized for events that might impact the original quality risk management decision, whether these events are planned (e.g., results ofproduct review, inspections, audits, change control) or unplanned (e.g., root cause from failure investigations, recall). The frequency of any review should be based upon the level of risk. Risk review might include reconsideration of risk acceptance decisions (section IV.D.4).
V. RISK MANAGEMENT METHODOLOGY (5)
Quality risk management supports a scientific and practical approach to decision making. It provides documented, transparent, and reproducible methods to accomplish steps of the quality risk management process based on current knowledge about assessing the probability, severity, and, sometimes, detectability of the risk.
Traditionally, risks to quality have been assessed and managed in a variety of informal ways (empirical and/or internal procedures) based on, for example, compilation of observations, trends, and other information. Such approaches continue to provide useful information that might support topics such as handling of complaints, quality defects, deviations, and allocation of resources.
In addition, the pharmaceutical industry and regulators can assess and manage risk using recognized risk management tools and/or internal procedures (e.g., standard operating procedures). Below is a nonexhaustive list of some of these tools (further details in Annex 1 and section VIII):
· Basic risk management facilitation methods
(flowcharts, check sheets, etc.)
· Failure Mode Effects Analysis (FMEA)
· Failure Mode, Effects, and Criticality Analysis (FMECA)
· Fault Tree Analysis (FTA)
· Hazard Analysis and Critical Control Points (HACCP)
· Hazard Operability Analysis (HAZOP)
· Preliminary Hazard Analysis (PHA)
· Risk ranking and filtering
· Supporting statistical tools
It might be appropriate to adapt these tools for use in specific areas pertaining to drug substance and drug productquality. Quality risk management methods and the supporting statistical tools can be used in combination (e.g., Probabilistic Risk Assessment). Combined use provides flexibility that can facilitate the application of quality risk management principles.
The degree of rigor and formality of quality risk management should reflect available knowledge and be commensurate with the complexity and/or criticality of the issue to be addressed.
VI. INTEGRATION OF QUALITY RISK MANAGEMENT INTO INDUSTRY AND REGULATORY OPERATIONS (6)
Quality risk management is a process that supports science-based and practical decisions when integrated into quality systems (see Annex II). As outlined in the introduction, appropriate use of quality risk management does not obviate industry’s obligation to comply with regulatory requirements. However, effective quality risk management can facilitate better and more informed decisions, can provide regulators with greater assurance of a company’s ability to deal with potential risks, and might affect the extent and level of direct regulatory oversight. In addition, quality risk management can facilitate better use of resources by all parties.
Training of both industry and regulatory personnel in quality risk management processes provides for greater understanding of decision-making processes and builds confidence in quality risk management outcomes.
Quality risk management should be integrated into existing operations and documented appropriately. Annex II provides examples of situations in which the use of the quality risk management process might provide information that could then be used in a variety of pharmaceutical operations. These examples are provided for illustrative purposes only and should not be considered a definitive or exhaustive list. These examples are not intended to create any new expectations beyond the requirements laid out in the current regulations.
Examples for industry and regulatory operations (see Annex II):
· Quality management
Examples for industry operations and activities (see Annex II):
· Development
· Facility, equipment, and utilities
· Materials management
· Production
· Laboratory control and stability testing
· Packaging and labeling
Examples for regulatory operations (see Annex II):
· Inspection and assessment activities
While regulatory decisions will continue to be taken on a regional basis, a common understanding and application of quality risk management principles could facilitate mutual confidence and promote more consistent decisions among regulators on the basis of the same information. This collaboration could be important in the development of policies and guidelines that integrate and support quality risk management practices.
VII. DEFINITIONS (7)
Decision maker(s): Person(s) with the competence and authority to make appropriate and timely quality risk management decisions.
Detectability: The ability to discover or determine the existence, presence, or fact of a hazard.
Harm: Damage to health, including the damage that can occur from loss of product quality or availability.
Hazard: The potential source of harm (ISO/IEC Guide 51).
Product lifecycle: All phases in the life of the product from the initial development through marketing until the product’s discontinuation.
Quality: The degree to which a set of inherent properties of a product, system, or process fulfills requirements (see ICH Q6A definition specifically for quality of drug substance and drug products).
Quality risk management: A systematic process for the assessment, control, communication, and review of risks to the quality of the drug product across the product lifecycle.
Quality system: The sum of all aspects of a system that implements quality policy and ensures that quality objectives are met.
Requirements: The explicit or implicit needs or expectations of the patients or their surrogates (e.g., health care professionals, regulators, and legislators). In this document, requirements refers not only to statutory, legislative, or regulatory requirements, but also to such needs and expectations.
Risk: The combination of the probability of occurrence of harm and the severity of that harm (ISO/IEC Guide 51).
Risk acceptance: The decision to accept risk (ISO Guide 73).
Risk analysis: The estimation of the risk associated with the identified hazards.
Risk assessment: A systematic process of organizing information to support a risk decision to be made within a risk management process. It consists of the identification of hazards and the analysis and evaluation of risks associated with exposure to those hazards.
Risk communication: The sharing of information about risk and risk management between the decision maker and other stakeholders.
Risk control: Actions implementing risk management decisions (ISO Guide 73).
Risk evaluation: The comparison of the estimated risk to given risk criteria using a quantitative or qualitative scale to determine the significance of the risk.
Risk identification: The systematic use of information to identify potential sources of harm (hazards) referring to the risk question or problem description.
Risk management: The systematic application of quality management policies, procedures, and practices to the tasks of assessing, controlling, communicating, and reviewing risk.
Risk reduction: Actions taken to lessen the probability of occurrence of harm and the severity of that harm.
Risk review: Review or monitoring of output/results of the risk management process considering (if appropriate) new knowledge and experience about the risk.
Severity: A measure of the possible consequences of a hazard.
Stakeholder: Any individual, group, or organization that can affect, be affected by, or perceive itself to be affected by a risk. Decision makers might also be stakeholders. For the purposes of this guidance, the primary stakeholders are the patient, healthcare professional, regulatory authority, and industry.
Trend: A statistical term referring to the direction or rate of change of a variable(s).
VIII. REFERENCES (8)
ICH Q8 Pharmaceutical Development.
ISO/IEC Guide 73:2002 - Risk management - Vocabulary - Guidelines for use in standards.
ISO/IEC Guide 51:1999 - Safety aspects - Guideline for their inclusion in standards.
Process Mapping, American Productivity & Quality Center, 2002, ISBN 1928593739.
IEC 61025 Fault tree analysis (FTA).
IEC 60812 Analysis techniques for system reliability—Procedure for failure mode and effects analysis (FMEA).
Failure Mode and Effect Analysis: FMEA from Theory to Execution, 2nd Edition 2003, D. H. Stamatis, ISBN 0873895983.
Guidelines for Failure Modes and Effects Analysis (FMEA) for Medical Devices, 2003, Dyadem Press, ISBN 0849319102.
The Basics of FMEA, Robin McDermott, Raymond J. Mikulak, Michael R. Beauregard, 1996, ISBN 0527763209.
WHO Technical Report Series No. 908, 2003, Annex 7 Application of Hazard Analysis and Critical Control Point (HACCP) methodology to pharmaceuticals.
IEC 61882 - Hazard Operability Analysis (HAZOP).
ISO 14971:2000 - Application of Risk Management to Medical Devices.
ISO 7870:1993 - Control Charts.
ISO 7871:1997 - Cumulative Sum Charts.
ISO 7966:1993 - Acceptance Control Charts.
ISO 8258:1991 - Shewhart Control Charts.
What is Total Quality Control?: The Japanese Way, Kaoru Ishikawa (Translated by David J. Liu, 1985, ISBN 0139524339.
ANNEX I: RISK MANAGEMENT METHODS AND TOOLS
The purpose of this annex is to provide a general overview of and references for some of the primary tools that might be used in quality risk management by industry and regulators. The references are included as an aid to gain more knowledge and detail about the particular tool. This is not an exhaustive list. It is important to note that no one tool or set of tools is applicable to every situation in which a quality risk management procedure is used.
I.1 Basic Risk Management Facilitation Methods
Some of the simple techniques that are commonly used to structure risk management by organizing data and facilitating decision making are:
· Flowcharts
· Check Sheets
· Process Mapping
· Cause and Effect Diagrams (also called an Ishikawa diagram or fish bone diagram)
I.2 Failure Mode Effects Analysis (FMEA)
FMEA (see IEC 60812) provides for an evaluation of potential failure modes for processes and their likely effect on outcomes and/or product performance. Once failure modes are established, risk reduction can be used to eliminate, contain, reduce, or control the potential failures. FMEA relies on product and process understanding. FMEA methodically breaks down the analysis of complex processes into manageable steps. It is a powerful tool for summarizing the important modes of failure, factors causing these failures, and the likely effects of these failures.
FMEA can be used to prioritize risks and monitor the effectiveness of risk control activities.
FMEA can be applied to equipment and facilities and might be used to analyze a manufacturing operation and its effect onproduct or process. It identifies elements/operations within the system that render it vulnerable. The output/results of FMEA can be used as a basis for design or further analysis or to guide resource deployment.
I.3 Failure Mode, Effects, and Criticality Analysis (FMECA)
FMEA might be extended to incorporate an investigation of the degree of severity of the consequences, their respective probabilities of occurrence, and their detectability, thereby becoming a Failure Mode, Effects, and Criticality Analysis (FMECA; see IEC 60812). In order for such an analysis to be performed, the product or process specifications should be established. FMECA can identify places where additional preventive actions might be appropriate to minimize risks.
Potential Areas of Use(s)
FMECA application in the pharmaceutical industry should mostly be utilized for failures and risks associated with manufacturing processes; however, it is not limited to this application. The output of an FMECA is a relative risk “score” for each failure mode, which is used to rank the modes on a relative risk basis.
I.4 Fault Tree Analysis (FTA)
The FTA tool (see IEC 61025) is an approach that assumes failure of the functionality of a product or process. This tool evaluates system (or subsystem) failures one at a time but can combine multiple causes of failure by identifying causal chains. The results are represented pictorially in the form of a tree of fault modes. At each level in the tree, combinations of fault modes are described with logical operators (AND, OR, etc.). FTA relies on the experts’ process understanding to identify causal factors.
Potential Areas of Use(s)
FTA can be used to establish the pathway to the root cause of the failure. FTA can be used to investigate complaints or deviations in order to fully understand their root cause and to ensure that intended improvements will fully resolve the issue and not lead to other issues (i.e. solve one problem yet cause a different problem). Fault Tree Analysis is an effective tool for evaluating how multiple factors affect a given issue. The output of an FTA includes a visual representation of failure modes. It is useful both for risk assessment and in developing monitoring programs.
I.5 Hazard Analysis and Critical Control Points (HACCP)
HACCP is a systematic, proactive, and preventive tool for assuring product quality, reliability, and safety (see WHO Technical Report Series No. 908, 2003, Annex 7). It is a structured approach that applies technical and scientific principles to analyze, evaluate, prevent, and control the risk or adverse consequence(s) of hazard(s) due to the design, development, production, and use of products.
HACCP consists of the following seven steps:
(1) conduct a hazard analysis and identify preventive measures for each step of the process
(2) determine the critical control points
(3) establish critical limits
(4) establish a system to monitor the critical control points
(5) establish the corrective action to be taken when monitoring indicates that the critical control points are not in a state of control
(6) establish system to verify that the HACCP system is working effectively
(7) establish a record-keeping system
Potential Areas of Use(s)
HACCP might be used to identify and manage risks associated with physical, chemical, and biological hazards (including microbiological contamination). HACCP is most useful when product and process understanding is sufficiently comprehensive to support identification of critical control points. The output of a HACCP analysis is risk management information that facilitates monitoring of critical points not only in the manufacturing process but also in other lifecycle phases.
I.6 Hazard Operability Analysis (HAZOP)
HAZOP (see IEC 61882) is based on a theory that assumes that risk events are caused by deviations from the design or operating intentions. It is a systematic brainstorming technique for identifying hazards using so-called guide words. Guide words (e.g., No, More, Other Than, Part of) are applied to relevant parameters (e.g., contamination, temperature) to help identify potential deviations from normal use or design intentions. HAZOP often uses a team of people with expertise covering the design of the process or product and its application.
Potential Areas of Use(s)
HAZOP can be applied to manufacturing processes, including outsourced production and formulation as well as the upstream suppliers, equipment and facilities for drug substances and drug products. It has also been used primarily in the pharmaceutical industry for evaluating process safety hazards. As is the case with HACCP, the output of a HAZOP analysis is a list of critical operations for risk management. This facilitates regular monitoring of critical points in the manufacturing process.
I.7 Preliminary Hazard Analysis (PHA)
PHA is a tool of analysis based on applying prior experience or knowledge of a hazard or failure to identify future hazards,hazardous situations and events that might cause harm, as well as to estimate their probability of occurrence for a given activity, facility, product, or system. The tool consists of: (1) the identification of the possibilities that the risk event happens, (2) the qualitative evaluation of the extent of possible injury or damage to health that could result, (3) a relative ranking of the hazard using a combination of severity and likelihood of occurrence, and (4) the identification of possible remedial measures
Potential Areas of Use(s)
PHA might be useful when analyzing existing systems or prioritizing hazards where circumstances prevent a more extensive technique from being used. It can be used for product, process and facility design as well as to evaluate the types of hazards for the general product type, then the product class, and finally the specific product. PHA is most commonly used early in the development of a project when there is little information on design details or operating procedures; thus, it will often be a precursor to further studies. Typically, hazards identified in the PHA are further assessed with other risk management tools such as those in this section.
I.8 Risk Ranking and Filtering
Risk ranking and filtering is a tool for comparing and ranking risks. Risk ranking of complex systems typically involves evaluation of multiple diverse quantitative and qualitative factors for each risk. The tool involves breaking down a basic risk question into as many components as needed to capture factors involved in the risk. These factors are combined into a single relative risk score that can then be used for ranking risks. “Filters,” in the form of weighting factors or cut-offs for risk scores, can be used to scale or fit the risk ranking to management or policy objectives.
Potential Areas of Use(s)
Risk ranking and filtering can be used to prioritize manufacturing sites for inspection/audit by regulators or industry. Risk ranking methods are particularly helpful in situations in which the portfolio of risks and the underlying consequences to be managed are diverse and difficult to compare using a single tool. Risk ranking is useful for management to evaluate both quantitatively-assessed and qualitatively-assessed risks within the same organizational framework.
I.9 Supporting Statistical Tools
Statistical tools can support and facilitate quality risk management. They can enable effective data assessment, aid in determining the significance of the data set(s), and facilitate more reliable decision making. A listing of some of the principal statistical tools commonly used in the pharmaceutical industry is provided:
l Control charts, for example:
— Acceptance control charts (see ISO 7966)
— Control charts with arithmetic average and warning limits (see ISO 7873)
— Cumulative sum charts (see ISO 7871)
— Shewhart control charts (see ISO 8258)
— Weighted moving average
l Design of experiments (DOE)
l Histograms
l Pareto charts
l Process capability analysis
ANNEX II: POTENTIAL APPLICATIONS FOR QUALITY RISK MANAGEMENT
This Annex is intended to identify potential uses of quality risk management principles and tools by industry and regulators. However, the selection of particular risk management tools is completely dependent upon specific facts and circumstances.
These examples are provided for illustrative purposes and only suggest potential uses of quality risk management. This Annex is not intended to create any new expectations beyond the current regulatory requirements.
II.1 Quality Risk Management as Part of Integrated Quality Management
Documentation
To review current interpretations and application of regulatory expectations
To determine the desirability of and/or develop the content for SOPs, guidances, etc.
Training and education
To determine the appropriateness of initial and/or ongoing training sessions based on education, experience, and working habits of staff, as well as on a periodic assessment of previous training (e.g., its effectiveness)
To identify the training, experience, qualifications, and physical abilities that allow personnel to perform an operation reliably and with no adverse impact on the quality of the product
Quality defects
To provide the basis for identifying, evaluating, and communicating the potential quality impact of a suspected quality defect, complaint, trend, deviation, investigation, out of specification result, etc.
To facilitate risk communications and determine appropriate action to address significant product defects, in conjunction with regulatory authorities (e.g., recall)
Auditing/Inspection
To define the frequency and scope of audits, both internal and external, taking into account factors such as:
· Existing legal requirements
· Overall compliance status and history of the company or facility
· Robustness of a company’s quality risk management activities
· Complexity of the site
· Complexity of the manufacturing process
· Complexity of the product and its therapeutic significance
· Number and significance of quality defects (e.g., recall)
· Results of previous audits/inspections
· Major changes of building, equipment, processes, key personnel
· Experience with manufacturing of a product (e.g., frequency, volume, number of batches)
· Test results of official control laboratories
Periodic review
To select, evaluate, and interpret trend results of data within the product quality review
To interpret monitoring data (e.g., to support an assessment of the appropriateness of revalidation or changes in sampling)
Change management/change control
To manage changes based on knowledge and information accumulated in pharmaceutical development and during manufacturing
To evaluate the impact of the changes on the availability of the final product
To evaluate the impact on product quality of changes to the facility, equipment, material, manufacturing process, or technical transfers
To determine appropriate actions preceding the implementation of a change, e.g., additional testing, (re)qualification, (re)validation, or communication with regulators
Continual improvement
To facilitate continual improvement in processes throughout the product lifecycle
II.2 Quality Risk Management as Part of Regulatory Operations
Inspection and assessment activities
To assist with resource allocation including, for example, inspection planning and frequency, and inspection and assessment intensity (see Auditing section in Annex II.1)
To evaluate the significance of, for example, quality defects, potential recalls, and inspectional findings
To determine the appropriateness and type of postinspection regulatory follow-up
To evaluate information submitted by industry, including pharmaceutical development information
To evaluate impact of proposed variations or changes
To identify risks that should be communicated between inspectors and assessors to facilitate better understanding of how risks can be or are controlled (e.g., parametric release, Process Analytical Technology (PAT)).
II.3 Quality Risk Management as Part of Development
To design a quality product and its manufacturing process to consistently deliver the intended performance of the product(see ICH Q8)
To enhance knowledge of product performance over a wide range of material attributes (e.g., particle size distribution, moisture content, flow properties), processing options, and process parameters
To assess the critical attributes of raw materials, solvents, active pharmaceutical ingredient (API) starting materials, APIs, excipients, or packaging materials
To establish appropriate specifications, identify critical process parameters, and establish manufacturing controls (e.g., using information from pharmaceutical development studies regarding the clinical significance of quality attributes and the ability to control them during processing)
To decrease variability of quality attributes:
· reduce product and material defects
· reduce manufacturing defects
To assess the need for additional studies (e.g., bioequivalence, stability) relating to scale up and technology transfer
To make use of the design space concept (see ICH Q8)
II.4 Quality Risk Management for Facilities, Equipment and Utilities
Design of facility/equipment
To determine appropriate zones when designing buildings and facilities, e.g.,
· flow of material and personnel
· minimize contamination
· pest control measures
· prevention of mix-ups
· open versus closed equipment
· clean rooms versus isolator technologies
· dedicated or segregated facilities/equipment
To determine appropriate product contact materials for equipment and containers (e.g., selection of stainless steel grade, gaskets, lubricants)
To determine appropriate utilities (e.g., steam; gases; power source; compressed air, heating, ventilation, and air conditioning (HVAC); water)
To determine appropriate preventive maintenance for associated equipment (e.g., inventory of necessary spare parts)
Hygiene aspects in facilities
To protect the product from environmental hazards, including chemical, microbiological, and physical hazards (e.g., determining appropriate clothing and gowning, hygiene concerns)
To protect the environment (e.g., personnel, potential for cross-contamination) from hazards related to the product being manufactured
Qualification of facility/equipment/utilities
To determine the scope and extent of qualification of facilities, buildings, and production equipment and/or laboratory instruments (including proper calibration methods)
Cleaning of equipment and environmental control
To differentiate efforts and decisions based on the intended use (e.g., multi- versus single-purpose, batch versus continuous production)
To determine acceptable (specified) cleaning validation limits
Calibration/preventive maintenance
To set appropriate calibration and maintenance schedules
Computer systems and computer-controlled equipment
To select the design of computer hardware and software (e.g., modular, structured, fault tolerance)
To determine the extent of validation, e.g.,
· identification of critical performance parameters
· selection of the requirements and design
· code review
· the extent of testing and test methods
· reliability of electronic records and signatures
II.5 Quality Risk Management as Part of Materials Management
Assessment and evaluation of suppliers and contract manufacturers
To provide a comprehensive evaluation of suppliers and contract manufacturers (e.g., auditing, supplier quality agreements)
Starting material
To assess differences and possible quality risks associated with variability in starting materials (e.g., age, route of synthesis).
Use of materials
To determine whether it is appropriate to use material under quarantine (e.g., for further internal processing)
To determine appropriateness of reprocessing, reworking, use of returned goods
Storage, logistics and distribution conditions
To assess the adequacy of arrangements to ensure maintenance of appropriate storage and transport conditions (e.g., temperature, humidity, container design)
To determine the effect on product quality of discrepancies in storage or transport conditions (e.g., cold chain management) in conjunction with other ICH guidances
To maintain infrastructure (e.g., capacity to ensure proper shipping conditions, interim storage, handling of hazardousmaterials and controlled substances, customs clearance)
To provide information for ensuring the availability of pharmaceuticals (e.g., ranking risks to the supply chain).
II.6 Quality Risk Management as Part of Production
Validation
To identify the scope and extent of verification, qualification, and validation activities (e.g., analytical methods, processes, equipment, and cleaning methods
To determine the extent for follow-up activities (e.g., sampling, monitoring, and re-validation)
To distinguish between critical and noncritical process steps to facilitate design of a validation study
In-process sampling & testing
To evaluate the frequency and extent of in-process control testing (e.g., to justify reduced testing under conditions of proven control)
To evaluate and justify the use of process analytical technologies (PAT) in conjunction with parametric and real time release
Production planning
To determine appropriate production planning (e.g., dedicated, campaign, and concurrent production process sequences)
II.7 Quality Risk Management as Part of Laboratory Control and Stability Studies
Out of specification results
To identify potential root causes and corrective actions during the investigation of out of specification results
Retest period/expiration date
To evaluate adequacy of storage and testing of intermediates, excipients, and starting materials
II.8 Quality Risk Management as Part of Packaging and Labeling
Design of packages
To design the secondary package for the protection of primary packaged product (e.g., to ensure product authenticity, label legibility)
Selection of container closure system
To determine the critical parameters of the container closure system
Label controls
To design label control procedures based on the potential for mix-ups involving different product labels, including different versions of the same label
[1] This guidance was developed within the Expert Working Group (Quality) of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) and has been subject to consultation by the regulatory parties, in accordance with the ICH process. This document has been endorsed by the ICH Steering Committee at Step 4 of the ICH process, November 2005. At Step 4 of the process, the final draft is recommended for adoption to the regulatory bodies of the European Union, Japan, and the United States.
[2] Arabic numbers reflect the organizational breakdown in the document endorsed by the ICH Steering Committee at Step 4 of the ICH process, November 2005.
![]() Back to Top
Back to Top ![]() Back to Guidance Page
Back to Guidance Page
![]() PDF requires the free Adobe Acrobat Reader
PDF requires the free Adobe Acrobat Reader
SOURCE















.jpg)






.jpg)

.jpg)









.jpg)

.jpg)














)
.jpg)


.jpg)








































































































































































































.jpg)

.jpg)











.jpg)


















































.jpg)















.jpg)






























































































































.jpg)

























