EU Technical File Compilation to Obtain CE Marking Approval
Emergo Group specializes in helping medical device and IVD companies prepare the necessary documentation to achieve CE Marking certification for their products. A required component is the CE Technical File (called a Design Dossier for Class III devices).

A CE Technical File is a comprehensive collection of information and documents that details everything about your medical device. Understanding how to assemble this information and present the information properly is essential. If you manufacture a Class I device, a less complex CE Marking Technical File may be required. In the case of Class IIa, IIb and Class III devices, a more complex CE Technical File or Design Dossier must be prepared.
EU Technical File construction is subject to review by a Notified Body if the medical device is Class I with measuring or sterile function, Class IIa, IIb, and III (Design Dossier). Once placed on the market, national Competent Authorities have the right to review EU Technical Files regardless of classification at any time. An incomplete or improperly completed Technical File may result in unexpected delays or even prevent market entry. To learn how the Technical File fits into the CE Marking process, watch our 7 minute Flash presentation that fully explains the European regulatory process (requires audio).
The components of a CE Mark Technical File include:
- Description of the product family and justification for why each product falls within the product family.
- Detailed account of the intended use of the device(s) including how the medical device(s) functions, what it does, where the device is used, what it is used with, and who uses it.
- Description of components, specifications, packaging and literature.
- The manufacturing process.
- Listing of accessories.
- Location of design responsibility and manufacturing facilities.
- Classification of the device and rationale for classification.
- The chosen route to compliance according to the applicable Directives.
- Declaration of Conformity that states the manufacturer's compliance with applicable Directive(s).
- Lifetime/shelf life of products and environmental limitations.
- Retention of QA, Competent Authority and Notified Body records.
- Vigilance reporting and Medical Device Reporting procedure.
- How and when to contact Competent Authorities.
- Name of, and contract with, your Authorized Representative in Europe.
- Subcontractor names and addresses, if applicable.
- Essential Requirements.
- Design input specifications.
- Application and references to Standards and Guidelines.
- Testing results and clinical evaluations.
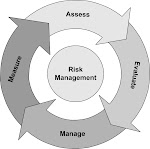
- Risk analysis.
- Instructions for Use and Labeling.
Emergo Group has compiled and reviewed CE Technical Files and Design Dossiers for hundreds of medical device and IVD companies. You can also read our in-depth article about the format and components of a Technical File.
As part of our CE Technical File or Design Dossier construction services, we will:
- Compile the EU Technical File or Design Dossier.
- Help you determine exactly which materials need to be assembled.
- Completely review all existing documentation in support of meeting the applicable Essential Requirements of the Directive(s).
- Determine applicable standards.
- Facilitate Risk Assessment.
- Review labeling and Instructions for Use.
- Develop and implement risk management process.
- Assist in writing the clinical evaluation summary.
Our European Technical File preparation services are handled on a fixed price basis.
Please contact us to learn more about our Technical File preparation services.
Source















.jpg)






.jpg)

.jpg)









.jpg)

.jpg)














)
.jpg)


.jpg)








































































































































































































.jpg)

.jpg)











.jpg)


















































.jpg)















.jpg)






























































































































.jpg)


























No comments:
Post a Comment